MAGIC
Recovering Gene Interactions from Single-Cell Data Using Data Diffusion

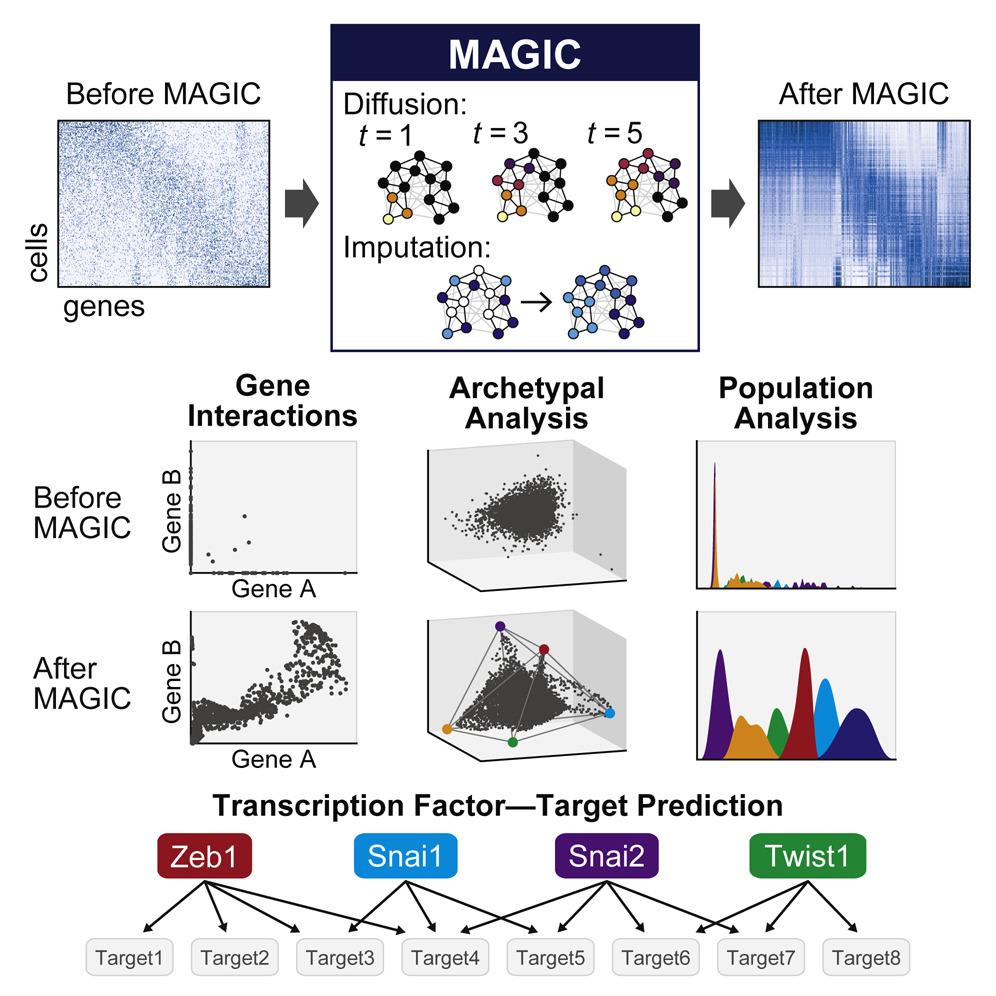
Single-cell RNA-sequencing is fast becoming a major technology that is revolutionizing biological discovery in fields such as development, immunology and cancer. The ability to simultaneously measure thousands of genes at single cell resolution allows, among other prospects, for the possibility of learning gene regulatory networks at large scales. However, scRNA-seq technologies suffer from many sources of significant technical noise, the most prominent of which is dropout due to inefficient mRNA capture. This results in data that has a high degree of sparsity, with typically only 10% non-zero values.
To address this, we developed MAGIC (Markov Affinity-based Graph Imputation of Cells), a method for imputing missing values, and restoring the structure of the data. After MAGIC, we find that two- and three-dimensional gene interactions are restored and that MAGIC is able to impute complex and non-linear shapes of interactions. MAGIC also retains cluster structure, enhances cluster-specific gene interactions and restores trajectories, as demonstrated in mouse retinal bipolar cells, hematopoiesis, and our newly generated epithelial-to-mesenchymal transition dataset.