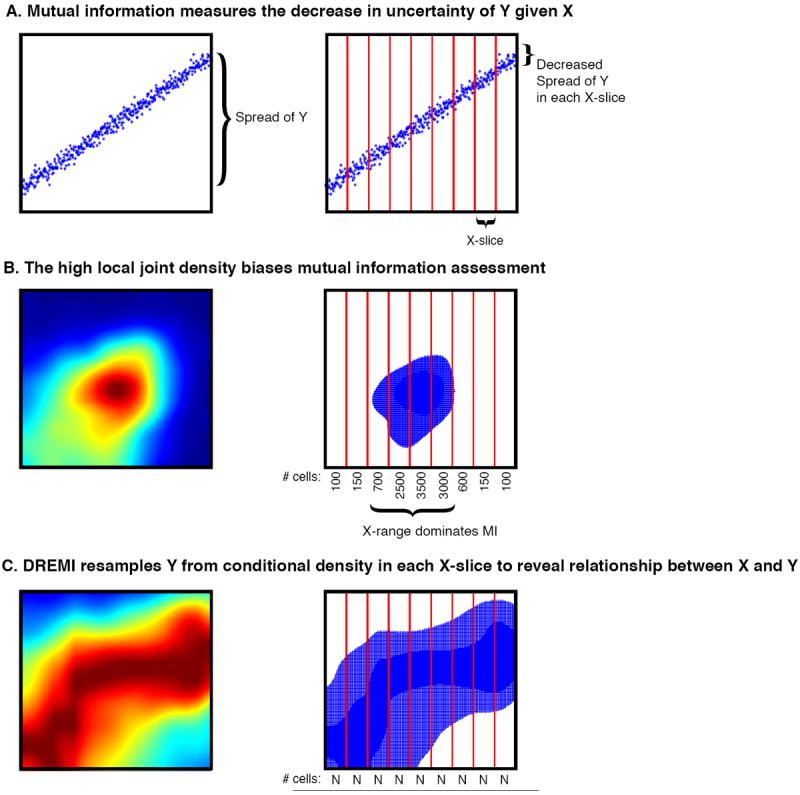
DREMI
Learning time-varying information flow from single-cell EMT data

Cellular circuits sense the environment, process signals, and compute decisions using networks of interacting proteins. To model such a system, the abundance of each activated protein species can be described as a stochastic function of the abundance of other proteins. High-dimensional single-cell technologies, like mass cytometry, offer an opportunity to characterize signaling circuit-wide. However, the challenge of developing and applying computational approaches to interpret such complex data remains.
Here, we developed computational methods, based on established statistical concepts, to characterize signaling network relationships by quantifying the strengths of network edges and deriving signaling response functions. In comparing signaling between naïve and antigen-exposed CD4+ T-lymphocytes, we find that although these two cell subtypes had similarly-wired networks, naïve cells transmitted more information along a key signaling cascade than did antigen-exposed cells.
We validated our characterization on mice lacking the extracellular-regulated MAP kinase (ERK2), which showed stronger influence of pERK on pS6 (phosphorylated-ribosomal protein S6), in naïve cells compared to antigen-exposed cells, as predicted. We demonstrate that by using cell-to-cell variation inherent in single cell data, we can algorithmically derive response functions underlying molecular circuits and drive the understanding of how cells process signals.